AlphaFold: Using AI for scientific discovery
Today we’re excited to share DeepMind’s first significant milestone in demonstrating how artificial intelligence research can drive and accelerate new scientific discoveries. With a strongly interdisciplinary approach to our work, DeepMind has brought together experts from the fields of structural biology, physics, and machine learning to apply cutting-edge techniques to predict the 3D structure of a protein based solely on its genetic sequence.
Our system, AlphaFold, which we have been working on for the past two years, builds on years of prior research in using vast genomic data to predict protein structure. The 3D models of proteins that AlphaFold generates are far more accurate than any that have come before—making significant progress on one of the core challenges in biology.
What is the protein folding problem?
Proteins are large, complex molecules essential in sustaining life. Nearly every function our body performs—contracting muscles, sensing light, or turning food into energy—can be traced back to one or more proteins and how they move and change. The recipes for those proteins—called genes—are encoded in our DNA.
What any given protein can do depends on its unique 3D structure. For example, antibody proteins that make up our immune systems are ‘Y-shaped’, and are akin to unique hooks. By latching on to viruses and bacteria, antibody proteins are able to detect and tag disease-causing microorganisms for extermination. Similarly, collagen proteins are shaped like cords, which transmit tension between cartilage, ligaments, bones, and skin. Other types of proteins include Cas9, which, using CRISPR sequences as a guide, act like scissors to cut and paste sections of DNA; antifreeze proteins, whose 3D structure allows them to bind to ice crystals and prevent organisms from freezing; and ribosomes that act like a programmed assembly line, which help build proteins themselves.
But figuring out the 3D shape of a protein purely from its genetic sequence is a complex task that scientists have found challenging for decades. The challenge is that DNA only contains information about the sequence of a protein’s building blocks called amino acid residues, which form long chains. Predicting how those chains will fold into the intricate 3D structure of a protein is what’s known as the “protein folding problem”.
The bigger the protein, the more complicated and difficult it is to model because there are more interactions between amino acids to take into account. As noted in Levinthal’s paradox, it would take longer than the age of the universe to enumerate all the possible configurations of a typical protein before reaching the right 3D structure.

Why is protein folding important?
The ability to predict a protein’s shape is useful to scientists because it is fundamental to understanding its role within the body, as well as diagnosing and treating diseases believed to be caused by misfolded proteins, such as Alzheimer’s, Parkinson’s, Huntington’s and cystic fibrosis.
We are especially excited about how it might improve our understanding of the body and how it works, enabling scientists to design new, effective cures for diseases more efficiently. As we acquire more knowledge about the shapes of proteins and how they operate through simulations and models, it opens up new potential within drug discovery while also reducing the costs associated with experimentation. That could ultimately improve the quality of life for millions of patients around the world.
An understanding of protein folding will also assist in protein design, which could unlock a tremendous number of benefits. For example, advances in biodegradable enzymes—which can be enabled by protein design—could help manage pollutants like plastic and oil, helping us break down waste in ways that are more friendly to our environment. In fact, researchers have already begun engineering bacteria to secrete proteins that will make waste biodegradable, and easier to process.
To catalyse research and measure progress on the newest methods for improving the accuracy of predictions, a biennial global competition called the Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP) was established in 1994, and has become the gold standard for assessing techniques.
How can AI make a difference?
Over the past five decades, scientists have been able to determine shapes of proteins in labs using experimental techniques like cryo-electron microscopy, nuclear magnetic resonance or X-ray crystallography, but each method depends on a lot of trial and error, which can take years and cost tens of thousands of dollars per structure. This is why biologists are turning to AI methods as an alternative to this long and laborious process for difficult proteins.
Fortunately, the field of genomics is quite rich in data thanks to the rapid reduction in the cost of genetic sequencing. As a result, deep learning approaches to the prediction problem that rely on genomic data have become increasingly popular in the last few years. DeepMind’s work on this problem resulted in AlphaFold, which we submitted to CASP this year. We’re proud to be part of what the CASP organisers have called “unprecedented progress in the ability of computational methods to predict protein structure,” placing first in rankings among the teams that entered (our entry is A7D).
Our team focused specifically on the hard problem of modelling target shapes from scratch, without using previously solved proteins as templates. We achieved a high degree of accuracy when predicting the physical properties of a protein structure, and then used two distinct methods to construct predictions of full protein structures.
Using neural networks to predict physical properties
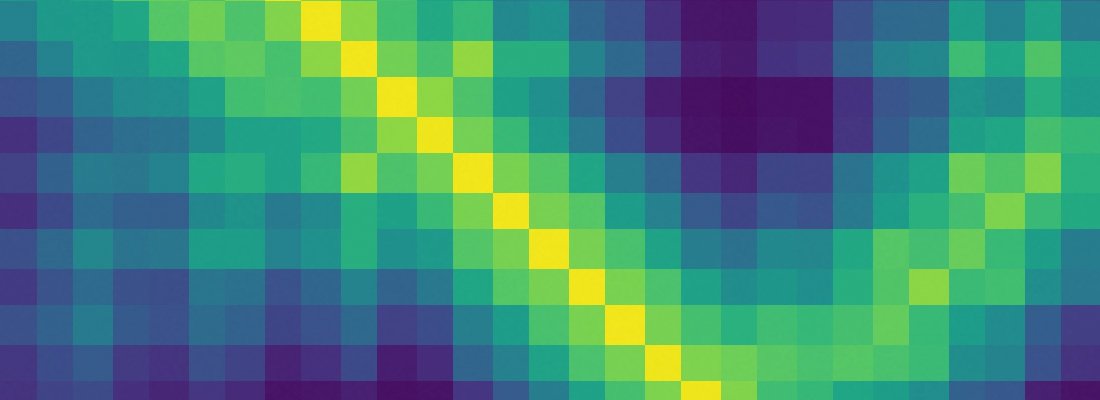
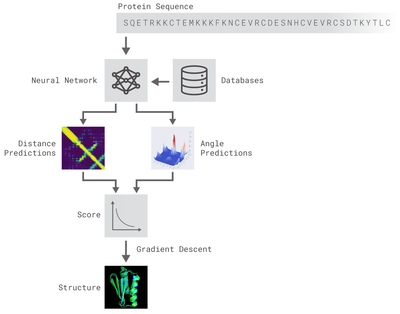
Both of these methods relied on deep neural networks that are trained to predict properties of the protein from its genetic sequence. The properties our networks predict are: (a) the distances between pairs of amino acids and (b) the angles between chemical bonds that connect those amino acids. The first development is an advance on commonly used techniques that estimate whether pairs of amino acids are near each other.
We trained a neural network to predict a separate distribution of distances between every pair of residues in a protein. These probabilities were then combined into a score that estimates how accurate a proposed protein structure is. We also trained a separate neural network that uses all distances in aggregate to estimate how close the proposed structure is to the right answer.
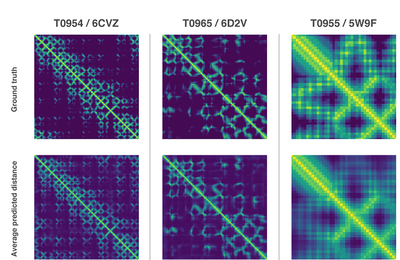
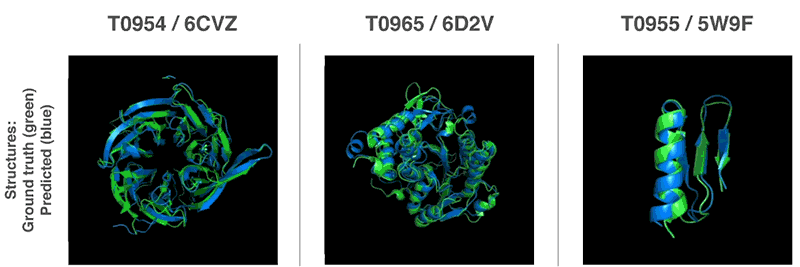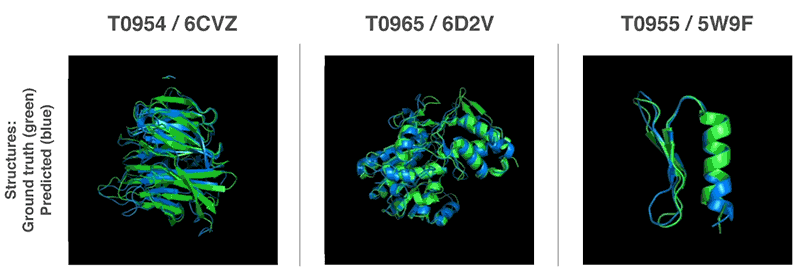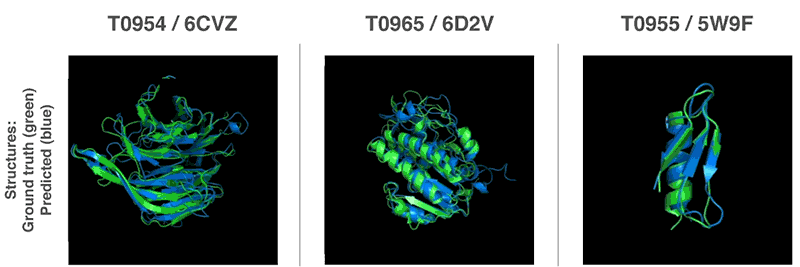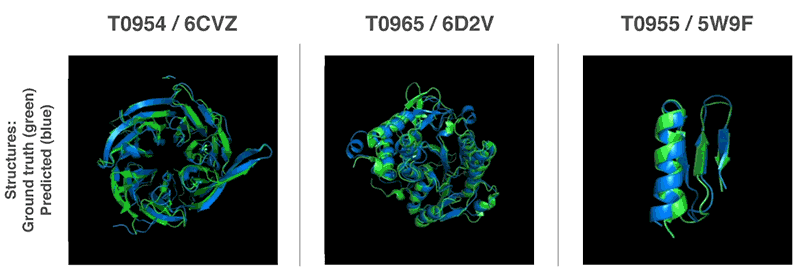
New methods to construct predictions of protein structures
Using these scoring functions, we were able to search the protein landscape to find structures that matched our predictions. Our first method built on techniques commonly used in structural biology, and repeatedly replaced pieces of a protein structure with new protein fragments. We trained a generative neural network to invent new fragments, which were used to continually improve the score of the proposed protein structure.
The second method optimised scores through gradient descent—a mathematical technique commonly used in machine learning for making small, incremental improvements—which resulted in highly accurate structures. This technique was applied to entire protein chains rather than to pieces that must be folded separately before being assembled, reducing the complexity of the prediction process.
What happens next?
The success of our first foray into protein folding is indicative of how machine learning systems can integrate diverse sources of information to help scientists come up with creative solutions to complex problems at speed. Just as we’ve seen how AI can help people master complex games through systems like AlphaGo and AlphaZero, we similarly hope that one day, AI breakthroughs will help us master fundamental scientific problems, too.
It’s exciting to see these early signs of progress in protein folding, demonstrating the utility of AI for scientific discovery. Even though there’s a lot more work to do before we’re able to have a quantifiable impact on treating diseases, managing the environment, and more, we know the potential is enormous. With a dedicated team focused on delving into how machine learning can advance the world of science, we’re looking forward to seeing the many ways our technology can make a difference.
Until we have published a paper on this work, please cite it as:
De novo structure prediction with deep-learning based scoring
R.Evans, J.Jumper, J.Kirkpatrick, L.Sifre, T.F.G.Green, C.Qin, A.Zidek, A.Nelson, A.Bridgland, H.Penedones, S.Petersen, K.Simonyan, S.Crossan, D.T.Jones, D.Silver, K.Kavukcuoglu, D.Hassabis, A.W.Senior
In Thirteenth Critical Assessment of Techniques for Protein Structure Prediction (Abstracts) 1-4 December 2018. Retrieved from here.
This work was done in collaboration with Richard Evans, John Jumper, James Kirkpatrick, Laurent Sifre, Tim Green, Chongli Qin, Augustin Zidek, Sandy Nelson, Alex Bridgland, Hugo Penedones, Stig Petersen, Karen Simonyan, Steve Crossan, David Jones, David Silver, Koray Kavukcuoglu, Demis Hassabis, and Andrew Senior